Birgit Strodel
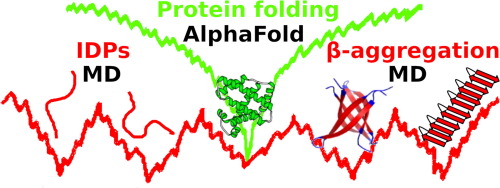
The protein folding problem was apparently solved recently by the advent of a deep learning method for protein structure prediction called AlphaFold. However, this program is not able to make predictions about the protein folding pathways. Moreover, it only treats about half of the human proteome, as the remaining proteins are intrinsically disordered or contain disordered regions. By definition these proteins differ from natively folded proteins and do not adopt a properly folded structure in solution. However these intrinsically disordered proteins (IDPs) also systematically differ in amino acid composition and uniquely often become folded upon binding to an interaction partner. These factors preclude solving IDP structures by current machine-learning methods like AlphaFold, which also cannot solve the protein aggregation problem, since this meta-folding process can give rise to different aggregate sizes and structures. An alternative computational method is provided by molecular dynamics simulations that already successfully explored the energy landscapes of IDP conformational switching and protein aggregation in multiple cases. These energy landscapes are very different from those of ‘simple’ protein folding, where one energy funnel leads to a unique protein structure. Instead, the energy landscapes of IDP conformational switching and protein aggregation feature a number of minima for different competing low-energy structures. In this review, I discuss the characteristics of these multifunneled energy landscapes in detail, illustrated by molecular dynamics simulations that elucidated the underlying conformational transitions and aggregation processes.